There was light snow this morning, but it has since melted away, leaving small puddles on the streets. Unfortunately, the sun seems to have lost today’s battle with the fog and the low clouds – it is, in essence, an ordinary wet, cold and gloomy February day. But if I peer out through the window, ignoring the construction works in the foreground, there is water on the horizon. And where there is water, there are ducks. And where there are ducks, there is flu. One cannot ask for more.
Over the years I have thought much about ducks and flu. (Some would say too much, but they don’t know what they miss). Although my research group has already produced four PhD theses on this topic, there is so much more that I would like to know. Some of it is highly specialized knowledge, of interest for a limited set of like-minded scientists with acquired duck disease tastes. Other things are quite basic, but hard to study, such as the question whether ducks infected with flu suffer from infection or not. That is a pretty important question also for a broader audience, as it has relevance for how well virus can spread with individuals in the environment; especially how ducks may spread virus long distances during migration. So, do they suffer from infections, or not?
Actually, there has been some controversy on this topic – partly stemming from different methods of quantifying disease effects. A field ecologist and a veterinarian have different scales in their toolboxes, one could say. In the latter case, disease signs are determined in experimental infections in animal house facilities, where individuals can be followed over time. Such experiments in Mallards have not been associated with strong disease signs – as long as we consider the low-pathogenic avian influenza viruses that are naturally occurring in wild avian populations (highly pathogenic AI is a completely different story). Infected Mallards shed viruses, but are otherwise apparently healthy, or only display a very short increase in body temperature. But, the ecologist argues, the artificial environment with plenty of food, controlled temperature and absence of predators is not really mimicking the situation in the wild, where even small reductions in vigilance and movement capacity may end in the death from a raptor’s claw. Absence of overt disease is not equal to absence of ecological costs, the ecologist would conclude.
The field studies so far have been a mixed bag, ranging from large effects to negligible effects depending on study and the species considered. The largest effect was seen in a study of Bewick’s swans in the Netherlands, where infected birds had poorer condition and migrated slower than uninfected swans. Such large effects have not been seen in other species, and one can not conclusively rule out other underlying factors, as the swan study was based on a limited number of birds. When it comes to Mallards – the most glorious of all avian influenza reservoir species – previous population studies from our group have suggested infected birds to weigh on average less than uninfected birds at capture.
Averages and populations are all and well, but to get to a mechanistic understanding one is better off with experiment conducted on a set of individuals. However, a problem is that we can not infect birds and release them in the field; in fact, we are not allowed to do so – there is a reason infection experiments are conducted in biosafety labs, after all. What to do, then? Well, we approached this question via GPS and accelerometer loggers attached to two groups of birds caught during the ongoing surveillance at our study site: one group of 20 Mallards with natural avian influenza infection at the time of capture, and another group of 20 Mallards that were negative for influenza at the time of capture.
The benefit of these data loggers is that they record such a wealth of information. From the GPS fixes we can follow the birds in the landscape and quantify their movements at spatial and temporal scales; from the accelerometer we can get metrics that describe activity, defined as movements in the x, y, z-dimensions. We predicted that infection would significantly hamper movement, and that with time the difference between infected and uninfected birds would level off (see figure below); hence the analyses need to take time in to account, too.

Theoretical predictions of the influence of infection on movement metrics. If infection affects spatial behaviour, infected (blue) and uninfected (red) birds should behave differently at the time of release. We postulate that, at this time, movement metrics for infected birds should be lower than for uninfected birds, which would be revealed as different intercepts of the regression of the movement metrics against time for uninfected (β0) and infected birds (β0+βInf). As infected birds recover with time, their movement metrics will approach and eventually meet the values for uninfected birds. This happens when the slope of the regression of the movement metrics against time for infected individuals (βT.aft.Rel*inf) reaches the slope for uninfected birds (βT.aft.Rel), which is expected to be null.
The full paper is freely accessible at Royal Society Open, and I hope readers with a more heavy interest in movement ecology download and read it. There is a lot of data crunching and statistics there that most of you are likely not that interested in – if you are, go read the original publication – but remember even easy questions may be hard to answer. Okay, with that said, what where the results?
Well. There were no effects of infection on the movement parameters measured, at all. Yes, there were differences among individuals, and between night and day, but infection status did not explain much of the variation in movement metrics. This means that under the natural situation in this study, conducted during stopover in autumn migration, infected ducks moved as much as uninfected ducks. This also means they likely are not impaired by infection during active migration, and could therefore carry LPAI viruses on the wing as they depart the stopover site.
Is this, then, the last nail in the ‘cost of infection’ coffin for low-pathogenic influenza in ducks? Probably not, because one could argue that non all viruses behave the same (in fact, there should be a variation for virulence), and that some viruses may have adapted to infect non-mallard-birds, and hence be spillover infections in Mallards (and then potentially be at less than optimal virulence). Moreover – and perhaps a stronger argument – there may be differences in outcome depending on whether it is a primary infection, or subsequent infection; where the first infection in a naïve bird could be believed to carry a larger cost. Or there may be effects seen only at certain environmental conditions.
All these ‘but, or, perhaps, mayhaps’ are classic scientist disclaimers… My personal belief, these days, is that also the ecological costs of infection are slim. But I am happy to be proven wrong – out you go now and study.
There is water at the horizon still. And questions aplenty.
Link to the article:
Bengtsson, D., Safi, K., Avril, A., Fiedler, W., Wikelski, M., Gunnarsson, G., Elmberg, J., Tolf, C., Olsen, B. & Waldenström, J. 2016. Does influenza A virus infection affect movement behaviour during stopover in its wild reservoir host? Royal Society Open Science 3: 150633.

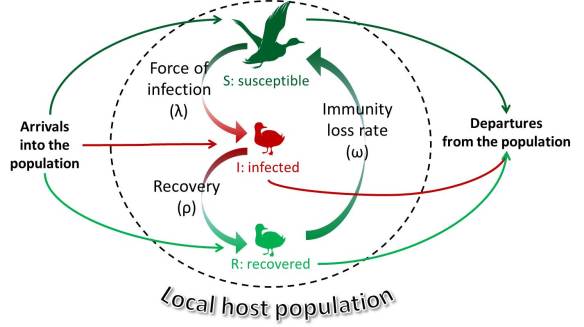
 For a pathogen to survive it has to find new hosts to infect. This may sound simple, but if you consider the entangled mesh that is the biology of a host species you realize that there are plenty of ways that things can go wrong, stopping the chain of transmission. First of all, the harm the pathogen incurs on its host – the virulence – needs to be balanced between being too low – the infection will be cleared before any transmission opportunities have occurred – or too high, so that it causes the demise of the host before transmission can take place. Second, the pathogens must overcome the hurdles of moving from one host to the next, be it in water, air or through the bite of an arthropod vector. And third, it has to overcome the fact that most hosts are not sedentary, but move varying distances in response to changes in the environment they inhabit. Finally, it needs to be able infect the new host and evade the immune system to establish infection. Not an easy feat, but something that is happening all the time in the world of viruses, bacteria, fungi, parasites and their hosts.
For a pathogen to survive it has to find new hosts to infect. This may sound simple, but if you consider the entangled mesh that is the biology of a host species you realize that there are plenty of ways that things can go wrong, stopping the chain of transmission. First of all, the harm the pathogen incurs on its host – the virulence – needs to be balanced between being too low – the infection will be cleared before any transmission opportunities have occurred – or too high, so that it causes the demise of the host before transmission can take place. Second, the pathogens must overcome the hurdles of moving from one host to the next, be it in water, air or through the bite of an arthropod vector. And third, it has to overcome the fact that most hosts are not sedentary, but move varying distances in response to changes in the environment they inhabit. Finally, it needs to be able infect the new host and evade the immune system to establish infection. Not an easy feat, but something that is happening all the time in the world of viruses, bacteria, fungi, parasites and their hosts. Hi there everyone, it is time for ducks and flu again! Just the other week we published a review on how host and virus traits affect transmission of low-pathogenic avian influenza viruses in wild birds. You should check it out – it is freely available in Current Opinion in Virology.
Hi there everyone, it is time for ducks and flu again! Just the other week we published a review on how host and virus traits affect transmission of low-pathogenic avian influenza viruses in wild birds. You should check it out – it is freely available in Current Opinion in Virology.